Press Release
The mathematics of repulsion for new graphene catalysts
ASSISTANT PROFESSOR. Tatsuhiko Ohto

Scientists at Tohoku University and colleagues in Japan have developed a mathematical model that abstracts the key effects of changes to the geometries of carbon material and predicts its unique properties. The details were published in the journal Carbon.
Scientists generally use mathematical models to predict the properties that might emerge when a material is changed in certain ways. Changing the geometry of three-dimensional (3D) graphene, which is made of networks of carbon atoms, by adding chemicals or introducing topological defects, can improve its catalytic properties, for example. But it has been difficult for scientists to understand why this happens exactly.
The new mathematical model, called standard realization with repulsive interaction (SRRI), reveals the relationship between these changes and the properties that arise from them. It does this using less computational power than the typical model employed for this purpose, called density functional theory (DFT), but it is less accurate.
With the SRRI model, the scientists have refined another existing model by showing the attractive and repulsive forces that exist between adjacent atoms in carbon-based materials. The SRRI model also takes into account two types of curvature in such materials: local curvatures and mean curvature.
The researchers, led by Tohoku University mathematician Motoko Kotani, used their model to predict the catalytic properties that would arise when local curvatures and dopants were introduced into 3D graphene. Their results were similar to those produced by the DFT model.
“The accuracy of the SRRI model showed a qualitative agreement with DFT calculations, and is able to screen through potential materials roughly one billion times faster than DFT,” says Kotani.
The team next fabricated the material and determined its properties using scanning electrochemical cell microscopy. This method can show a direct link between the material’s geometry and its catalytic activity. It revealed that the catalytically active sites are on the local curvatures.
“Our mathematical model can be used as an effective pre-screening tool for exploring new 2D and 3D carbon materials for unique properties before applying DFT modelling,” says Kotani. “This shows the importance of mathematics in accelerating material design.”
The team next plans to use their model to look for links between the design of a material and its mechanical and electron transport properties.
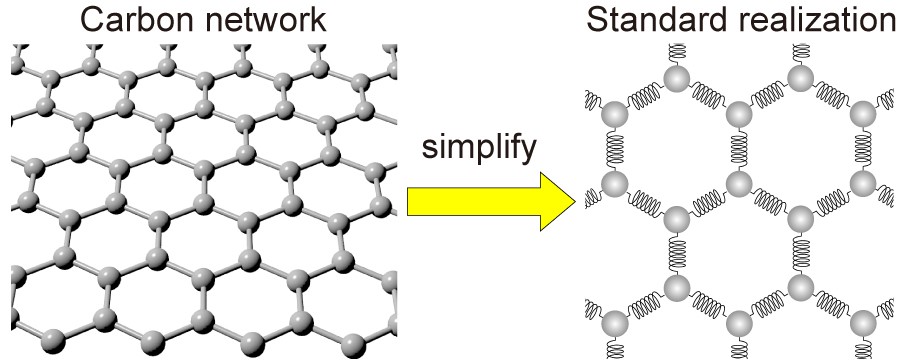
The original ‘standard realization’ model simplifies what a carbon network looks like, replacing the atoms with balls and the bonds with springs. The new modified model describes the attractive and repulsive forces between the carbon atoms.
Please see ResOU for the detail.